
![]()
![]()
![]()
![]()
![]()
![]()
![]()
|
For The Agency | For The Technology Developer | For The Magazine Publisher | For The Individual |
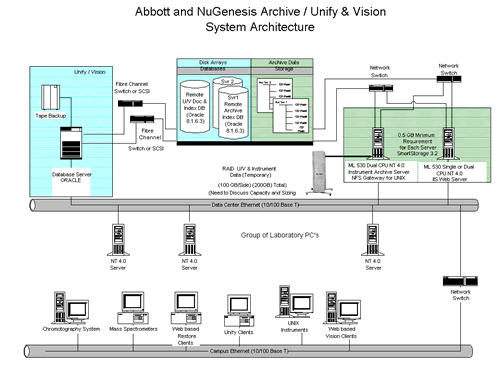
Software Helps Pharma Companies Meet FDA RulesThe FDA has stepped up its enforcement of 21 CFR Part 11 rules regarding electronic signatures and document standards. Developers of electronic documentation software are helping pharmaceuticals companies meet this challenge. The following is a manuscript for an article published in R&D magazine. R&D magazine holds the copyright for the finished article. C.G. Masi, Contributing Editor "Pharmaceuticals companies now have to relate all of their islands of information into their ocean of information," says Wolfgang Winter, Product Manager for Data Systems for the Life Science Business Unit of Agilent Technologies, Waldbronn, Germany. "We are talking about networking all of these DAQ systems so that they report all of their data into a central data repository. Fig. 1 shows a typical pharmaceutical-enterprise data system. Sources for new electronic documents include automated instrumentation systems, laboratory PCs (used to generate reports, memos, etc.), and archive database servers (used to metaanalyze historical records as well as archiving new documents). All these records now have to be generated, stored and modified in a 21 CFR Part 11 compliant way.
With the combination of GLP (good laboratory practice) rules and the new rule on electronic signatures (21 CFR Part 11), the question of instrument control, especially the question of trust-worthy and reliable instrument control, becomes a very tricky thing. Data is being generated in the form of electronic records. These electronic records fall under the requirements of 21 CFR 11 Part 11. Now, the question is: "What measures are in place in a data system or data acquisition system that make instrument control reliable and trustworthy and traceable." In a nutshell, Part 11 requests technical controls that maximize the chances of detecting falsifications if they should ever occur intentionally or by mistake, and minimize the chances of introducing any falsifications. The regulation intends to make electronic records trustworthy and reliable on top of making individuals accountable for what they do to the data. It's similar to signing a contract or a check with your hand-written signature. Part 11 just establishes the technical background that allows electronic records and signatures to be treated the equivalent of hand-signed documents on paper. "There really are benefits to moving to a clean-record world," Mary Ellen Goffredo, Vice-President of Marketing, NuGenesis Technologies Corporation, Westborough, Mass. points out. "If you take a step back and look at the consequences of Part 11 from a business point of view, you find that there are real benefits towards deploying electronic systems. Benefits include increased speed of information exchange, reducing errors in data movement and transcription, better data integration, improved process control. "There are also economies involved," Goffredo adds, "because paper takes up a lot of physical room." "The only people today that don't have to worry about Part 11 compliance are people that are in early drug discovery phases," says Mark Harnois, Senior Product Marketing Manager for Waters Corporation, Milford, Mass. "but, a lot of those people are very interested in this regulation, anyway." The big motivation for deploying Part 11 compliant systems in early drug discovery is to establish a legal basis for patent applications. Researchers have to be able to identify when discoveries were made, who made them, etc. What's the Rush? In the past, the FDA has been rather lenient about 21 CFR Part11 compliance. Indications are that this honeymoon is over. If you're information systems have not been made compliant yet, you need to do something about it immediately. "21 CFR Part 11 went into effect in August of 1997 and it said that legacy systems needed to be brought into compliance," Harnois points out. "We're now in 2001 and there are still a number of systems that are generating electronic records that are not compliant. Back in 1997, there were no systems in place that were compliant, but now in 2001 there are systems in place that could make your operation compliant. When an auditor comes in, they're going to want to see that you've made progress toward that goal." In the regulation, the FDA never stated a particular deadline. The indicators that you should be looking at are the frequency of warning letters that are coming up that are specific to Part 11. Back in 1999, the number of warning letters is estimated at about two. The estimate for 2000 is about 15. There have been about 20 just in this first half of 2001. Enforcement is clearly accelerating."You didn't see the FDA enforcing the regulation in full force until approximately a year ago," Harnois reports. "That is when the warning letters started to come out." "Over the last 18 months in particular, it's become quite apparent that Part 11 here to stay," Goffredo echos. "Many of our clients are now telling us that they perceive Part 11 compliance to be a bigger task than Y2K. Not only bigger in terms of the scope of the project but also bigger in terms of expense." History DJ: "I think it all started with the paperwork reduction act," says David Janelle, the Vice President of Systems Development for Doxis, Norwood, Mass. "The Federal Government wanted to reduce paperwork within Federal agencies. The FDA, in particular, was just being inundated with paper. Pharmaceutical companies also wanted to be able to submit information electronically. But, before that could happen, there had to be rules and regulations governing those records." "The FDA never really recommends any technical specs," Winter points out. "They just spell out a very abstract formulation of the requirements. It's up to the industry to interpret them and come up with suitable solutions." "21 CFR Part 11 sits on top of other regulations," explains Harnois. "It says if the other regulations that you're following in the conduct of your process require you to generate electronic records, then you have to follow these additional regulations." "Those records are required by what are called the predicate rules" Goffredo adds. "They are the GMP (good manufacturing practice), GLP (good laboratory practice), GCP (good clinical practice) regulations. if you generate those records electronically, then Part 11 applies. Part 11 only refers to records that are required by these predicate rules" This interpretation causes most of the data systems used by pharmaceuticals companies to fall under the Part 11. Examples include: chromatography data systems, LIMS systems, automated document-management systems, batch records, standard operating procedures, inventory records, calibration systems, preventative-maintenance records, training records, customer complaint files, and adverse-event reporting systems. These are all records that are used to meet those predicate rule requirements, so they can be covered by Part 11. "When electronic records are required," says Harnois, "you must comply with the regulations that are set forth for electronic records. "Electronic signatures, on the other hand, are not required; they are optional. You don't need to incorporate electronic signatures into your system. You can maintain written signatures. However, for efficiency and for performance and for work flow, we see more and more companies moving in that direction." "What you have to think about is what kind of processes exist in the lab that require people to sign something on paper," Winter points out. "All Part 11 is trying to do is rebuild these paper processes in the electronic world. So, if there is any aspect of your paper process that is being rebuilt or modeled in a software application, you have to think about a system for signing off electronically." One of the ways that you could do that would be to use the operating system, such as Windows NT, to secure the signatures. That would be considered somewhat of a hybrid system--hybrid meaning that you're using some components of an operating system, and some components of your application to secure the application." "The problem is that Windows NT wasn't designed to comply with Part 11," says Harnois. "It was never written to do that. It was really written to secure a network. It has some functions that can help, but it's not going to have all that are needed." Making Systems Compliant "No software product is compliant by itself," Harnois points out. "No matter what the product can do, a compliant system is going to include a combination of your administrative and procedural controls along with the technical controls that are built into the software." Furthermore, the FDA will never come and audit a company for Part 11 compliance. What they do is look at your drug development and manufacturing process. If they find that you are doing functions that generate electronic records, they will start to ask questions about your electronic documentation security. You can't, of course, make your entire data system compliant instantaneously. What the FDA wants is to see a plan and some progress toward implementing that plan. Table 1 lists the stages any 21 CFR Part 11 compliance program has to go through. The first step is assessing your documentation systems for Part 11 compliance, which is called a gap analysis. You evaluate the entire system with an eye toward Part 11 compliance. Once you have identified where the problems are, the second step is to put procedural or administrative controls in place to fill the gaps temporarily. Examples of procedural controls are standard operating procedures that say:
The third step is to deploy compliant-ready software systems that minimize your need for procedural controls through the use of technical controls. Technical controls are functions built into the software running your data systems that force users into Part-11-compliant behavior. The best way to see examples of these technical controls is to look at examples of compliant-ready software that is now available. Instrumentation Software "We have all the technical controls built into our Millennium software," Waters' Harnois says, "so that you can deploy it in a compliant fashion. But it's still the responsibility of the customer. The FDA will never come and audit a software vendor. They only audit the people that produce the pharmaceutical products." Millennium has been around for over 10 years. From the very beginning has included an embedded Oracle database, built-in security functions and an audit-trail mechanism. Early versions, however, really wouldn't pass muster as the basis for a 21 CFR Part 11 compliant data acquisition system. There were still too many gaps that users would have to fill with procedural controls. The latest Millennium version (3.2), which was introduced soon after the regulation went into effect, filled the most important of those gaps. It is, therefore, the company's first truly compliant-ready product. The company's goal is to provide more and more technical controls to eliminate the need for procedural and administrative controls and make the process of deployment as simple as possible. An important compliance feature is called the "System Policies Page." It provides a series of check boxes that system administrators can use to implement their procedural controls. For example, there is a check box that, when selected, tells the software require a minimum password length. "Our customers can walk up to the software and, just by selecting a series of check boxes, make their system Part 11 compliant. For example, each person will be identified by a user name and a password. Associated with that identity is a user type, which determines what that person has the ability to do and not do in the system. If the administrator assigns a "chemist" user type, that person can collect data, process data, but I can't modify reports. Someone with a "manager" user type can modify things or delete things, but not collect raw data. The definitions of what a "chemist" should be able to do versus what a "supervisor" or a "manager" can do are all part of the setup for your particular installation. In general, it will be the system administrator who defines the security set up in a given installation as a procedural control. Another interesting feature appears in the audit trail function. A basic tenet of Part 11 compliance is that you can't overwrite a file. Most non-compliant software systems allow overwriting files in order to save disk space. Some software packages attempt to provide a compliant audit trail by keeping a log file telling you what changes were made, who made them and when they made them. "Our software," says Harnois, "doesn't overwrite a file. You can go back and look at anything you've ever created. You can visually review the product from cradle to grave." Data Management "Our goal is to make all of the disparate data and data types used in a pharmaceuticals enterprise come together in a single central repository," says NuGenesis' Goffredo. "That allows users to reduce administrative costs and helps people collaborate throughout their company." NuGenesis' philosophy is that such a scientific data management system (SDMS) should be automatic; it should be accurate; it should be independent of whatever application was used to originally create the data; it should also be non-invasive and able to scale across the enterprise. It should be based on industry standards, run on common operating systems, such as Windows and UNIX, use a standard database management system like Oracle and employ thin-client web-browser technology. It should also be easy to use and easy to deploy. Finally, and very importantly, it should comply with 21 CFR Part 11 in terms of security, audit trails, and access privileges. "Our NuGenesis Scientific Data Management System (SDMS) allows you to collect all different kinds of data in a single Oracle database," Goffredo continues. "We have a unique set of tools that allow you to get data into these Oracle databases in an unattended, automated noninvasive fashion. The end-user scientist doesn't have to change anything that they do today to get their data into one of the NuGenesis databases." The NuGenesis SDMS allows you to capture data from any source. Part 11 says that you need to make complete and accurate copies of both the machine-readable and human-readable data, so the system is designed to do that. After capturing that data, the system catalogues it in an Oracle database. The company's proprietary technology allows extracting relevant metadata to aid subsequent retrieval. Finally, the system is web enabled, so that you can find that information instantly via your enterprise-wide network for analysis and review, incorporate it into FDA presentations, and so forth. For example, consider data acquired using an HPLC controlled by Waters' Millennium system described above. In chromatography you'll make an injection into an HPLC system, it will generate a chromatogram and the raw output from the HPLC will go into the chromatography data system. the chromatography data system will apply an integration algorithm, to figure out how big the peaks are. That algorithm will tie into a calibration table, which will tell you what a specific peak area means. Finally, the system will put the result into a specific format based on a report method. If that particular record is required by GMP rules, the FDA can ask to see the record and all of the raw data that created that record. So, if two years from now the FDA wants to know what Charlie did on Friday afternoon, 15 June, the company needs to be able to recreate that report in electronic form. In order to do that it needs to have the binary data from the detector, the integration method, the calibration method and the report method, and then be able to reprocess it all and recreate the report. NuGenesis' SDMS allows you to find all of the data on demand. You can search and retrieve data within minutes, whereas before it might have taken days or weeks. From a Part 11 perspective, web-based tools are wonderful because they minimize the code that has to run on the client systems. Having web-based software allows you to distribute new tools to thin clients throughout the enterprise without having to do complete revalidation of existing systems, which can save millions of dollars. Networked Data Systems Thin-client web technology is an important part of Agilent's data-system philosophy as well. They also feel that it makes deploying Part 11-compliant systems easier. The company's Cerity system, a member of the Agilent Family of Networked Data Systems, is targeted at Pharmaceutical QA/QC labs, and models the way people work in a pharmaceutical QA/QC environment. The networked data systems control multitechnique instrumentation--combinations of liquid chromatographs (LCs), gas chromatographs (GCs), mass spectrometers (MSs) and other spectrometers, etc. There are also general-purpose interfaces that allow users to capture digital output from just about any other device that exists as well. The networked data system collects data from the various instrumentation systems in a pharmaceutical company's labs. It can also control any instrument that is suitably equipped for remote control. It interprets the signals and spits out the numeric results upon which some kind of decision will be made. These instruments can all be connected to a central data repository, which is an Oracle database that pulls together all of the data that is being measured in the analytical lab. That is useful not just for archival purposes and for backup, but for also for correlation putting pieces of data together that have been measured over time on different instruments by different people. Attached to every record, there are globally unique identifiers (GUIDs) linked to the individual who performed the tests. Agilent lets the operating system to handle security functions, such as password authentication. "The system administrator takes care of making sure the logons are unique and ensures that there is an appropriate password policy is in place. "All of that is handled beautifully by Windows NT," Winter reports. Software to Help Develop Your Compliant System "We looked at the rules pharmaceutical companies follow as far as filling out paper," says Doxis' Janelle. "For example, to make a change, you use a single-line strikeout with an initial and a date. If somebody comes along and makes a change to that piece of paper much later, they also sign it." One of the challenges Doxis had when designing their system was to develop software that allowed users to follow that same signoff policy while ensuring that nobody could make a change to the information directly in the database outside the application. "We force users to interact with the data in a Part 11 compliant manner," he says. There are several components that make up the application. The three main components of Doxis' system are:
Authoring Component The Authoring application allows a nonprogrammer to create a form using Microsoft Word skills. These forms become the online screens users interact with when performing laboratory operations. For example, to run a sample through a GC, the user must either define a method or call up a predefined method that will control the instrument during the test. GLP requires that this method become part of the documentation for the test run. Part 11 then sets the standard for that document's security, including control of who can fill out the forms in order to run the test. Once created, these forms become electronic records as well, so they fall under Part 11 requirements. That means you have to have approvals and signoffs for creating them as well as filling them out, and they all have to be under revision control so that nobody can change those documents without authorization. Administration Component As with the other software systems described, the system administrator needs a tool to designate who should be allowed to do what and when. The administrator uses Doxis Administration to add a user, give them a user name, a password, and designate their sign-off privileges. Through a series of check boxes, the system administrator designates the new user as a "technician," so all he does is fill out forms as part of operating the equipment. The next person might be a technician and an author, so they can design forms as well as fill out forms. Fill-IT Component The Doxis system controls what users can do while filling out a form. Users, for example, can only go from intelligent field to intelligent field. They can't change any of the other (fixed) information on the form. Fill-IT forces the user to follow all of the rules that the author built into it. For example, if the author specified a range of 5 to 15 for a certain field, and the user tries to put in 16, /fill-IT will reject it. If the author said that the first two characters in another field have to be alpha and the next five have to be numeric, that's all Fill-IT will accept. All of these controls are there so an auditor can go back, look at each field and see when each entry was filled out and make sure that the form was signed (by putting in the user's ID and pass-word) subsequent to filling out all the information. SOURCES Mark Harnois, Senior Product Marketing Manager for Waters Corporation, Milford, Massachusetts Mary Ellen Goffredo, Vice-President of Marketing, NuGenesis Technologies Corporation, Westborough, Mass. ngoffredo@nugenesis.com Wolfgang Winter, Product Manager for Data Systems for the Life Science Business Unit of Agilent Technologies, Waldbronn, Germany David Janelle, the Vice President of Systems Development for Doxis, Norwood, Mass. Table 1: Typical steps to achieving 21 CFR Part 11 compliance
For More Information |
|
Home | About Us | Technology Journalism | Technology Trends Library | Online Resources | Contact Us For The Agency | For The Technology Developer | For The Magazine Publisher | For The Individual © , C. G. Masi Technology Communications, Privacy Policy P.O. Box 10640, 978 S. San Pedro Road, Golden Valley, AZ 86413, USA Phone: +1 928.565.4514, Fax: +1 928.565.4533, Email: cgmasi@cgmasi.com, Web: www.cgmasi.com Developed by Telesian Technology Inc. |